We are happy to announce the new jMRUI version 7.0
This new version comes loaded with many improvements and new features, amongst the most important:
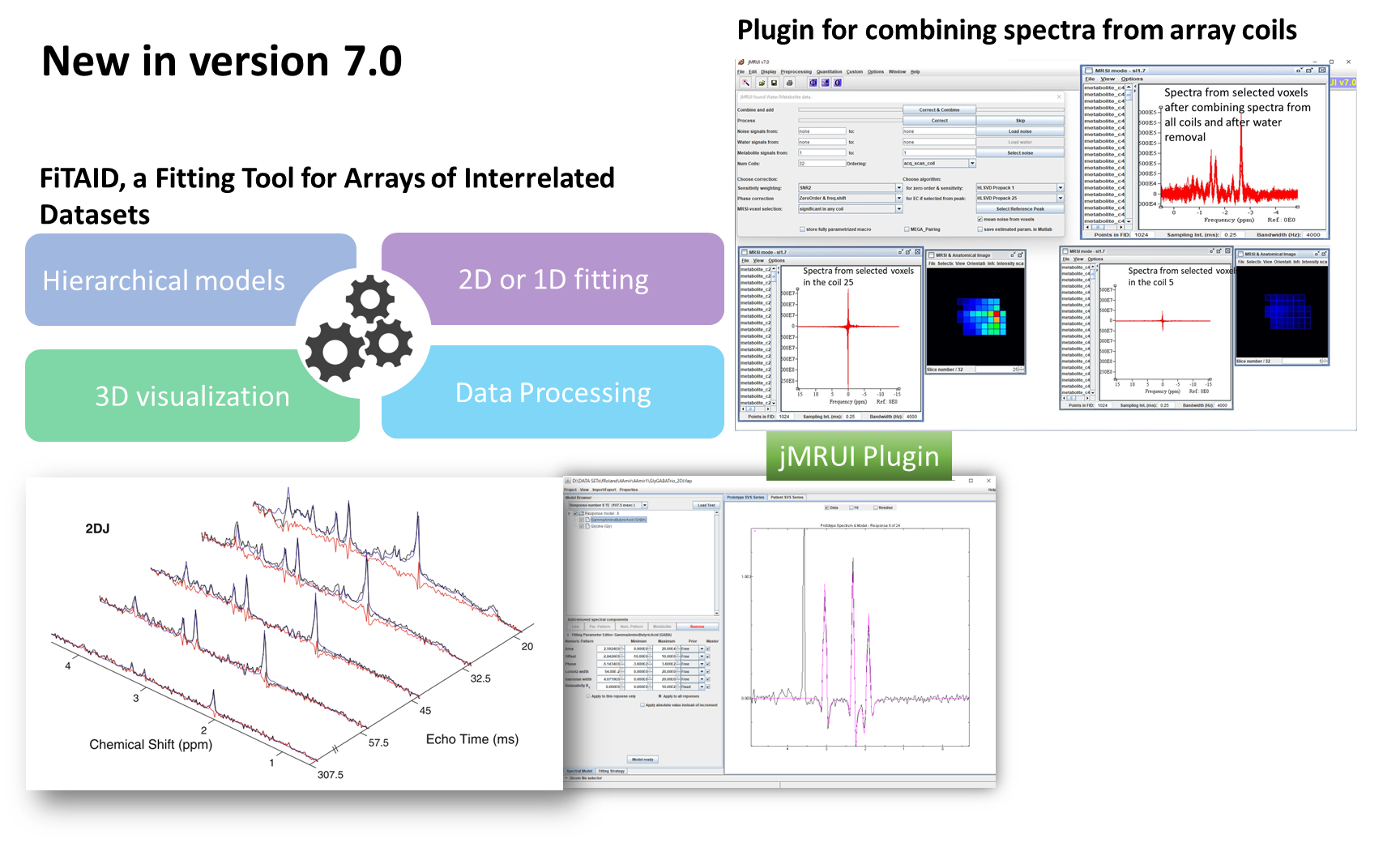
- FiTAID – Fitting Tool for Arrays of Interrelated Datasets. A new plugin for multidimensional, interrelated spectra quantification: models for 1D, 2D and multidimensional fitting (Saturation recovery, Inversion recovery, 2DJ, 2DJ diffusion, 2DJIR); flexible construction of the signal model with metabolite profiles defined as combinations of simulated signal profile(s) (simulated, e.g., in VeSPA, NMRScopeB (jMRUI)) and/or resonance peaks (Gaussian, Lorentzian, Voigt shapes); possibility to set various constraints on estimated parameters; possibility to select the fitting strategy (e.g. fitting domain, simultaneous or individual fit, algorithm, fitting region etc.); visualization tools for multidimensional data; batch processing of large multi-dimensional MRS data;
- Flexible algorithm for combining spectra from array coils, tested for GE and Bruker raw data
- plugin for phase and frequency correction of SV or MRSI spectra, based on a selected peak;
- improved handling and processing of multi-slice MRSI data;
- QUASARY (improved QUASARX), i.e., QUASAR with a choice of Gaussian or Lorentzian lineshape, common damping for selected metabolites, fixed amplitude ratios between selected metabolites;
- AMARES is now accessible from the plugin SpectrIm and the resulting metabolite maps obtained in AMARES can be imported back into SpectrIm for visualization; the AMARES starting values, peak positions, and other prior knowledge can be saved/loaded as text files with a single click;
- NMRScopeB implemented with Python3, with improved protocols;
- support of new data formats: Bruker PV360, new GE data format, MRS-NIfTI data standard format in jMRUI; option to store/load quantification results and simulated spectra in JSON/NIfTI format; spectra including MRSI data can be saved in Matlab; Philips Advanced DICOM integrated in SpectrIm.




